

El
análisis del antiguo ADN afirma que el cánido de Altai como un
perro primitivo.
El
origen de los perros domésticos sigue siendo polémica, con los
datos genéticos que indica una separación entre los perros y los
lobos modernos en el Pleistoceno tardío. Sin embargo, sólo unos
pocos como perros fósiles se encuentran antes del Último Máximo
Glacial, y es ampliamente aceptado que la domesticación del perro es
anterior al inicio de la agricultura hace unos 10.000 años.
Con
el fin de evaluar la relación genética de una de las más antiguas
de los perros, se ha aislado el antiguo ADN de la recientemente
descrita de un perro putativo Pleistoceno de 33.000 años de edad, de
Altai y se analizaron 413 nucleótidos de la región de control
mitocondrial. Nuestro análisis revela que el haplotipo único del
perro de Altai está más estrechamente relacionado con los perros
modernos y cánidos prehistóricos del Nuevo Mundo de lo que es para
los lobos actuales. Otros análisis genéticos de los cánidos
antiguos puede revelar una fecha más exacta y el centro de la
domesticación.
La
domesticación de los perros del lobo gris es bien aceptada. Sin
embargo, el momento y el lugar y el número de eventos de
domesticación sigue siendo activamente debatido. El registro
arqueológico proporciona que el perro sigue siendo inequívoco
comenzando hace unos 14.000 años de calendario (CY) y que requieren
una domesticación que es anterior a la agricultura.

Los
presuntos perros permanece con edades comprendidas entre 31.000 a
36.000 cy, han sido cuestionados como potencialmente representan
intentos abortados de domesticación, o lobos morfológicamente
únicos. Un análisis completo del genoma mitocondrial de los perros
modernos sugiere un origen en el sur de China alrededor de hace
16.000 años, mientras que un amplio nuclear del genoma en todo el
análisis de SNP apoya un origen de Oriente Medio y Europa, que está
más de acuerdo con los datos arqueológicos .
Aquí
hemos aislado, secuenciado y analizado 413 nucleótidos de la región
de control del ADN mitocondrial de un espécimen de perro putativo de
una fecha como de aproximada. 33.000 cy de las montañas de Altai en
Asia Central. Sólo se sabe un espécimen único, el perro de Goyet
(36.000 cy) que es anterior al perro Altai y por lo tanto es hasta
ahora el segundo espécimen más antiguo conocido asignado
morfológicamente al perro doméstico.
Materiales
y Métodos
La
muestra
El
cráneo de un antiguo perro de 33.000 cy, como cánido utilizado en
este estudio fue desenterrado en Razboinichya Cave (montañas de
Altai del sur de Siberia, Figura 1), en 1975, como se describe
anteriormente. La excavación fue autorizado plenamente por las
autoridades gubernamentales y llevada a cabo en virtud de
subvenciones correspondientes de la Academia Rusa de las Ciencias. En
la actualidad, la muestra es parte de la colección del Instituto de
Arqueología y Etnografía SB RAS. Dientes y fragmentos mandibulares
de esta muestra fueron proporcionadas por el Dr. ND Ovodov, coautor y
miembro de este Instituto, que tenía autorización institucional
para proporcionar este material.
La
extracción del ADN, amplificación y secuenciación
Se
extrajo el ADN de un incisivo derecho lateral inferior y un fragmento
de hueso mandibular del cánido parecido a un perro. Hemos llevado a
cabo dos extracciones independientes del antiguo ADN a lo descrito
previamente, con algunas modificaciones. Todas las etapas de
extracción seguida de estrictos criterios necesarios para garantizar
la autenticidad del antiguo ADN, tales como trabajar en un
laboratorio independiente, aislado y con medidas para evitar y
detectar la contaminación incluidos los controles negativos (por
ejemplo, mezcla de extracción sin ningún material óseo) .
La
capa de la superficie del hueso (0.5-1.0 mm) se eliminó con un
taladro y el material óseo interior se molió en un mortero hasta
obtener un polvo fino. De 1 a 1,5 g del polvo se volvió a suspender
en 15 ml de 0,5 M EDTA pH 8,0, 0,5% de N-lauril sarcosil
(Sigma-Aldrich, Alemania) y 0,5 mg / ml de proteinasa K. La
suspensión se incubó durante 15 min a 55 ° C. Las piezas sin
disolver se separaron por centrifugación a 5.000 g durante 5 min.
La
primera fracción se eliminó y los elementos sólidos restantes se
incubaron de nuevo a 55 º C hasta que todo el material óseo se
disolvió. El sobrenadante se concentró mediante Amicon Ultra-15
concentrador (Millipore, Alemania) con un tamaño de exclusión de 5
kDa hasta un volumen de 100-120 l. El producto fue purificado
utilizando el QIAquick PCR Purification Kit (Qiagen, Alemania) según
las instrucciones del fabricante.

El
antiguo ADN recién aislado fue amplificado utilizando la
amplificación de todo el genoma (WGA) Kit (Sigma-Aldrich, Alemania)
como se ha descrito previamente. Hemos aplicado este método para
aumentar la cantidad inicial del aDNA con el fin de que esté
disponible para posteriores experimentos. Los cebadores para la
amplificación de la región de control mitocondrial del antiguo
perro se diseñaron basándose en un análisis publicados
previamente. Sin embargo, hemos ampliado la longitud de los cebadores
con base en la secuencia del ADN mitocondrial de Canis familiaris
(perro EU789784) y Canis lupus (lobo FJ978035) a partir de GenBank,
por lo que podría aumentar la temperatura de hibridación de la PCR.
Los
cebadores se enumeran en la Tabla S1. La localización de los
cebadores con respecto a EU789784 secuencia de referencia GenBank se
muestra en la Figura S1. La longitud de producto de PCR varió desde
170 hasta 389 pb. Para evitar la amplificación de las citosinas
desaminados comunes en el antiguo ADN, se utilizó el Phusion de alta
fidelidad de la ADN polimerasa (Thermo Scientific, Finlandia)
siguiendo las instrucciones del fabricante. Para cada PCR se
emplearon concentraciones estándar de tampón (fabricante incluido),
dNTPs (0,2 mM), cebadores (1 M) y enzima (0,02 U / l). Una l de la
ADN (WGA producto) se añadió a la 25 l de la mezcla de reacción.
El
protocolo de PCR incluía 35 ciclos de 3 min a 98 ° C, 30 seg a 70 °
C, y 10 seg a 72 ° C. Después de dos rondas de amplificación por
PCR, los residuos de PCR se separaron por electroforesis en gel y se
separa por escisión de banda. El ADN de la banda que contiene los
productos de la PCR se aisló utilizando el Gel Extraction Kit
(Qiagen, Alemania). La secuenciación con los mismos cebadores usados
para la amplificación se llevó a cabo en el centro
Interinstitucional de secuenciación de ADN en SB RAS.

Los
análisis de datos
La
secuencia se comparó primero a las secuencias públicamente
disponibles en la base de datos NCBI nr por medio de búsquedas
BLASTN. Con el fin de realizar la asignación de probabilidad, la
red, y la reconstrucción filogenética de árboles, incorporamos la
secuencia de la muestra de Altai en una alineación de 72 perros (70
razas conocidas), 30 lobos y coyotes (cuatro Tabla S2). Además de
los perros y los lobos actuales, que además incluye información de
la secuencia de 35 especímenes prehistóricos de cánidos del nuevo
mundo.
Se
evaluó el modelo de sustitución más adecuado para nuestro conjunto
de datos mediante la opción de prueba del modelo implementado en el
programa MEGA 5. Hemos aplicado para que sea el mejor modelo o bien
apoyado alternativas siempre que sea el modelo más probable en el
que no estaba disponible en los programas que se utilizan para la
reconstrucción filogenética.
El
mapeo de probabilidad, permite la visualización de informatividad
filogenético de un alineamiento de secuencia. Utilizando una
ponderación de un cuarteto de topología, este método proporciona
una poderosa herramienta para evaluar si la secuencia de evolución
se produjo de una manera en forma de estrella o el resultado de un
árbol completamente resuelto. Además, predefiniendo la secuencia de
las agrupaciones, el método puede ser utilizado para evaluar el
apoyo de cada una de las posibles topologías en un cuarteto.
En
este caso, se realizó un mapa de Probabilidad utilizando TREE-PUZZLE
v5.2. Todos los cuartetos posibles se utilizaron cuando pueden
inferir en el apoyo de diferentes topologías para los arreglos de
ramas diferentes. Sin embargo, al evaluar el conjunto de los datos
completo sin particiones, sólo 10.000 cuartetos fueron aplicados. El
HKY con cinco categorías de tasa de gamma fue utilizado como un
modelo de sustitución.
 |
| Figura S 2 |
El
programa de la red v4.610 se utilizó para construir una red
filogenético de todos los haplotipos. Una ventaja de esta
reconstrucción filogenética es una visualización completa de las
relaciones de haplotipos complejos mediante la creación de una red
en lugar de un árbol y que incluye los posibles nodos ancestrales o
intermedio. Con el fin de reducir la complejidad de la red, los
haplotipos idénticos se combinaron y sus respectivas frecuencias se
indican con el tamaño de los círculos en la figura S3.
Hemos
ajustado la tasa de transición / transversion dando cinco veces más
peso a transversions y enraizado en la red con los coyotes. Se
combinaron el algoritmo mediana uniendose con la opción de MP para
reducir aún más la complejidad de la red producidas por exclusión
de los enlaces superfluos.
Por
último, hemos utilizado el genoma mitocondrial completo de todos los
perros y los lobos contemporáneos con el fin de mantener grupos
estadísticamente bien compatibles de todos los perros y los lobos
(véase la figura S4) y se evaluó la posición relativa de la
muestra de Altai por medio de la conexión vecina, máxima parsimonia
y máxima reconstrucciones de árboles de probabilidad que emplean
MEGA 5. Con el fin de evaluar la solidez y el apoyo estadístico de
los árboles, que corrió cada análisis filogenético con 1.000
bootstraps pero variaba el árbol de algoritmos de búsqueda, así
como los modelos de sustitución.
Las distancias genéticas se
calcularon bajo el supuesto de los parámetros Kimura 2, el modelo de
mejor ajuste cuando se aplica una alineación truncada de 413
nucleótidos, y las pruebas de significación estadística mediante
la prueba de Mann-Whitney U implementado en un paquete R-software.
 |
| Fifura S 3 |
Resultados
y Argumento
Se
obtuvieron las secuencias de ADN mitocondrial de la región de
control tanto de los dientes y la mandíbula de la muestra de 33.000
cy del antiguo perro putativo de Altai y nos parecieron ser
idénticos. Con el fin de evaluar la relación genética de la
muestra de Altai a cualquier conocida muestra de perro / lobo, hemos
realizado varios análisis. En primer lugar, una búsqueda BLASTN de
la secuencia de Altai (413 nucleótidos) contra la base de datos NCBI
nr de manifiesto la similitud en el nivel de 99% (puntuación máx.
756), pero no es rival perfecto para cualquier perro o lobo
existente. Puesto que la secuencia que se encontró era única, fue
depositada en GenBank con el número de acceso JX173682.
En
segundo lugar, comparamos nuestra secuencia de tres ya publicadas de
57-nucleótidos fragmentos de ADN mitocondrial del lobo de la misma
cueva (cueva Razboinichya, 32.500, 48.000 y 50.000 años sin
calibrar,) y se encontró al perro de Altai de ser diferente a los 2,
3 y 3 posiciones de nucleótidos, respectivamente. Esto indica que
los lobos anteriormente descritos del pleistoceno de la cueva
Razboinichya no están estrechamente relacionados con la muestra
estudiada aquí. Sin embargo, más datos de los lobos prehistóricos
de la misma región se necesitan para estimar la diversidad de la
población y obtener una visión más completa de las relaciones
genéticas de los cánidos Altai.
Las
Reconstrucciones filogenéticas inequívocas de la historia evolutiva
de los perros y los lobos contemporáneos han sido a menudo
obstaculizado por eventos de hibridación dentro del género Canis,
lo que lleva a los árboles sin resolver o valores bajos de apoyo de
los patrones de ramificación cuando se utilizó la información
mitocondrial. Al investigar la informatividad filogenética de
nuestra base de datos que combina la muestra de Altai, 72 perros
existentes, 30 lobos y 35 cánidos prehistóricos del Nuevo Mundo y
cuatro coyotes también encontramos poco apoyo ya sea para un patrón
claramente resuelto ramificación o de evolución en forma de
estrella (Figura S2).
 |
| Tabla S 2 |
El
patrón de distribución de los cuartetos de 10.000 investigados
(Figura S2A) reveló una probabilidad igual de tres topologías
resueltas (23,5%, 23,8% y 23,7% en la Figura S2B) y sólo una
probabilidad ligeramente menor para una evolución en forma de
estrella en la que los tres topologías tienen la misma probabilidad
(21,8%). Este resultado se apoya además en las redes de haplotipos
que se manifestaron en forma de estrella en patrones de divergencia
(por ejemplo H_1 (incluyendo la muestra de Altai), H_48 o H_2 (Figura
S3)).
Strimmer
y Haeseler sugieren que el análisis de secuencias más largas puede
aumentar la informatividad de la alineación y, en consecuencia, se
compara entonces la secuencia de Altai para el genoma mitocondrial
completo de 72 perros, 30 lobos y cuatro coyotes como así como las
secuencias superpuestas de un total de 35 precolombinos perros y
cánidos del Pleistoceno. La opción de la eliminación por pares
aplicados en el análisis filogenético nos permitió retener el
agrupamiento haplotipo basada en genomas mitocondriales completos de
perros contemporáneos (Figura 2, véase también la figura S4,
Figura S5), y para colocar el haplotipo Altai con respecto a los
subtipos de perros grandes . El vecino a participado en árbol
bootstrap de secuencias antiguas y modernas que muestra una
separación bien apoyada de todos los perros contemporáneos de
coyotes y dos haplotipos de lobo basal (Figura 2).
El
haplotipo derivado de los grupos de muestras de Altai entre las
secuencias obtenidas a partir de las pre-colombinas de los perros y
otros cánidos del Pleistoceno Superior como el lobo. Este grupo de
haplotipos se incrusta en un clado que comprende secuencias de perros
exclusivamente contemporáneos (Clado A) y contiene la mayoría de
los haplotipos del perro incluyendo las razas tan diversas como el
mastín tibetano, el Terranova, el Crestado Chino, Cocker Spaniel o
Siberian Husky (Tabla S2). Cabe destacar que el apoyo estadístico de
los análisis filogenéticos es débil ya que los valores de arranque
son bajos (por debajo de 50 valores se omiten pero véase la Figura
S5).
 |
| Figura S 5 |
Sin
embargo, a pesar de la disposición de los haplotipos individuales
cambian cada vez que la aplicación de diferentes métodos de
construcción de árboles (máxima verosimilitud, máxima parsimonia,
participación del Vecino), la relación cercana y la colocación del
Altai haplotipos dentro del perro Clade A eran consecuentes. Esta
relación se ve apoyada por los análisis adicionales, tales como una
asignación de probabilidad de agrupamiento de cuatro (Figura 3) o la
red de haplotipos (Figura S3).
La
asignación de la probabilidad revela un fuerte apoyo para una
topología de agrupar la muestra de Altai con perros contemporáneos
(Figura 3A: 53,9%) a favor de los acuerdos de agrupación de la
muestra Altai, ya sea con coyotes o lobos (Figura 3A: 1,6%, 8,4%,
respectivamente). Cuando se intercambian los coyotes con cánidos
prehistóricos del Nuevo Mundo a partir de las topologías que une la
muestra de Altai con perros contemporáneos o cánidos prehistóricos
eran casi igualmente probables. Sin embargo, estas asociaciones eran
aproximadamente cuatro veces más probable que una disposición de la
muestra de Altai con los lobos modernos.
Una
inspección más a fondo de la red haplotipo también revela que los
haplotipos que tienen la relación más estrecha con la muestra de
Altai (incrustados en H_1 en la Figura S3) se componen del perro o
prehistoricos cánidos haplotipos del Nuevo Mundo (Figura S3, Tabla
S3). Con el fin de evaluar adicionalmente estas relaciones estrechas
se analizó la distancia genética pairwise, asumiendo un modelo de
sustitución del Parámetro de Kimura 2 (Figura 4) y se encontró
que la agrupación haplotipos en Clado A, tenía la menor distancia
genética (2-5 diferencias sobre una alineación de 413 nucleótidos
), seguido por las distancias más pequeñas que existen en las
segundas diferentes comparaciones de los perros pre-colombinos, y la
mayor distancia de las diferencias a los lobos actuales. Además, al
comparar las distancias derivadas de todos los haplotipos de los
perros para que de todos los lobos, encontramos una distancia
significativamente más pequeña genética de la muestra de Altai a
los perros modernos (Mann-Whitney U, p <0 font="">

En
conclusión, nuestros análisis apoyan la hipótesis de que la
muestra de Altai que está más estrechamente relacionada con los
perros domésticos que a los lobos existentes, pero hacemos hincapié
en el punto de que estos análisis se limita a un solo lugar, de
herencia materna y que se necesitaría más datos de la secuencia
para obtener una filogenia estadísticamente bien apoyada y sin
ambigüedades para resolver la relación genética de la muestra de
Altai. Sin embargo, este análisis preliminar confirma la conclusión
de que la muestra de Altai que es probable que un perro antiguo con
una divergencia superficial de los lobos antiguos. Estos resultados
sugieren una historia más antigua del perro que fuera de Oriente
Medio o de Asia oriental, como ya se sugirió de los centros del
origen del perro. Los descubrimientos adicionales de los antiguos
restos como del perro son esenciales para seguir reduciendo el tiempo
y la región de origen del perro doméstico.
Agradecimientos
Nos
gustaría dar las gracias al editor y dos revisores por sus
comentarios constructivos que mejoraron el manuscrito. OT agradece a
D. Schwochow por sus valiosos comentarios sobre una versión anterior
del manuscrito.
Contribuciones
de los autores
Concebido
y diseñado los experimentos: RKW ASG. Realizó los experimentos:
ASD. Analizados los datos: ASD OT. Contribuido reactivos / materiales
/ herramientas de análisis: NO NVV. Escribió el documento: OT IVA
ASD JAL RKW ASG.
Por:
Anna S. Druzhkova equal contributor, Olaf Thalmann equal contributor,
Vladimir A. Trifonov mail, Jennifer A. Leonard, Nadezhda V.
Vorobieva, Nikolai D. Ovodov, Alexander S. Graphodatsky, Robert K.
Wayne
Tradu
y Publi: Erik Farina, Etólogo Canino, Psicolmascot.
 |
| Figura 1. Mapa que muestra el origen geográfico de la muestra del perro de Altai. |

Figura
2 : Árbol Consenso de vecinos Participar (1.000 pasos de
arranque), construido suponiendo que el modelo de sustitución de
Tamura-Nei, el mejor modelo de ajuste para el conjunto de datos que
comprende genomas mitocondriales completos de coyotes (Coyotes),
lobos (OWW, NWW - lobos del Viejo Mundo y el Nuevo, respectivamente)
y perros combinados con secuencias parciales de la región de control
de la muestra Altai (perro de Altai) y otros cánidos prehistóricos
(perros pre-colombinos, lobos orientales Beringia). Destacamos todos
los clados que contienen perros modernos en Clado luz azul y ampliada
A para una mejor visibilidad. La posición de la muestra de Altai
está marcado con una flecha de luz azul en la ampliación. Bootstrap
valores se muestran con un asterisco siempre mayor que 50.

Figura
3. Probabilidad del análisis mapeado de los 142 secuencias
cánidos agrupados en cuatro grupos. El panel superior muestra el
patrón de distribución de todos los cuartetos y el panel inferior
representa la fracción de cada región ocupada. Cuartetos situadas
en la parte central del triángulo compatible con una secuencia de la
evolución en forma de estrella mientras que los cuartetos en las
tres esquinas de apoyo decidido topologías, respectivamente. A) del
patrón de asignación de probabilidad cuando la agrupación de las
secuencias del perro de Altai, perros, lobos y coyotes. B) el patrón
de asignación de la probabilidad cuando las secuencias se agruparon
de la siguiente manera: Perro de Altai, los perros, los lobos y
cánidos prehistóricos del Nuevo Mundo.
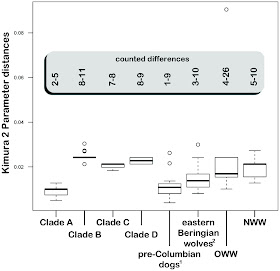
Figura
4. Diagrama de caja que muestra las diferencias genéticas entre
la muestra de Altai y varias agrupaciones de cánidos actuales y
extintos. Las distancias genéticas se calcularon bajo el supuesto
modelo de sustitución de parámetros de Kimura 2, el mejor ajuste
para la alineación truncado de los cánidos. La caja resaltada
muestra las diferencias mínimas y máximas contadas. 1 Leonard et
al. 2007, 2 Leonard et al. 2002.

Figura
S1. El esquema de secuenciación del perro de Altai superpone a
la secuencia de ADN mitocondrial canino de GenBank (EU789784). Las
líneas verticales discontinuas indican los límites de 413 pb de
secuencia utilizado en este trabajo. Las flechas pequeñas indican
las posiciones de todos los cebadores de la Tabla S1. Bares 1-6
indican reacciones de PCR independientes con diferentes combinaciones
de cebadores: (1) - D1F/D1R (365 pb), (2) - D2F/D09R (389 pb), (3) -
D1F/D2R (343 pb); (4) - D10F/D09R (195 pb), (5) - D3F/D3R (212 pb),
(6) - D5F/D09R (170 pb).

Figura
S2. Probabilidad del análisis de mapeo de todos los cánidos 142
sin compartimentación adicional de los datos. El panel superior
muestra el patrón de la distribución de todos los cuartetos y el
panel inferior representa la fracción de cada región ocupada.

Figura
S3. Red Haplotipo que resumen
las relaciones filogenéticas de los haplotipos únicos. Haplotipos
idénticos que se colapsó y los tamaños de los círculos indican
las frecuencias. Haplotipos diferentes están etiquetados con H_XX
(círculos amarillos) y la hipótesis de los vectores de la mediana
con mvXX (círculos rojos). La longitud de los enlaces entre los
nodos es proporcional a las diferencias mutacionales. Para una mejor
visibilidad, el enlace a la raíz (coyotes) y dos anómalos
haplotipos lobo se truncan y el grupo de haplotipos que contiene el
perro Altai está resaltado en verde.

Figura
S4. Árbol de los vecinos que
Participan generado con los genomas mitocondriales de 72 perros y los
30 lobos con 1.000 pasos de arranque. Las áreas sombreadas indican
los cuatro grupos bien apoyados del perro y el punto de flechas a los
valores de soporte para cada clado.

Figura
S5. Los Vecinos que Participan
en el árbol que representa una versión totalmente anotada del árbol
que se muestra en la Figura 2 generado con 413 pb de la región
hipervariable del genoma mitocondrial. Los valores en rojo indican el
apoyo de arranque después de 1.000 pasos. Los identificadores se
explica en la Tabla S2 con la excepción de las secuencias
etiquetadas "JAL". La nomenclatura se adopta este último
de Leonard et al. 2007 [24] y Leonard et al. 2002 [23] y las flechas
apuntan a los valores de soporte para cada clado.
 |
Tabla
S1. Lista de los cebadores
utilizados en este estudio.
|
 |
Tabla
S2. Nomenclatura, número de
acceso y raza / origen geográfico de los distintos haplotipos del
perro / lobo.
|
 |
Tabla
S3. Asignaciones de haplotipos
para el análisis de redes.
|
Tradu
y Publi: Erik Farina, Etólogo Canino, Psicolmascot.
Contacto:
psicolmascot@gmail.com